DNASTAR 作为分子生物学领域的核心软件套件,广泛应用于基因序列分析、蛋白质结构预测及多组学研究。其灵活的序列管理功能是科研人员高效处理数据的基石。本文从基础操作到高阶应用,详细解析DNASTAR 怎么导入序列以及DNASTAR 怎么加载序列的核心步骤,助力用户快速掌握关键技术,提升实验数据处理效率。
一、DNASTAR 怎么导入序列

导入序列是DNASTAR 进行数据分析的第一步,支持从本地文件、数据库或在线资源直接加载数据。以下为详细操作流程及注意事项:
1.本地文件导入
支持格式:DNASTAR 兼容FASTA、Gen Bank(.gb)、EMBL(.embl)、ABI(.ab1)等主流格式;
操作步骤:
打开DNASTAR 软件,点击菜单栏“File”→“Import”→“Sequence File”;
在弹出窗口中选择目标文件(可多选批量导入),勾选“自动识别文件编码”(Auto-DetectEncoding);
针对ABI测序文件,需额外勾选“解析质量评分”(ParseQualityScores)以保留原始测序信噪比数据;
高级配置:
在“Import Options”中设置默认序列类型(DNA/RNA/Protein),避免后续手动分类;
启用“去除低质量区段”(TrimLow-Quality Regions),设定Phred评分阈值(建议≥20)。
2.数据库直接调用
通过“Database”模块连接NCBIGenBank或Uni Prot数据库:
输入序列名称或ID(如NM_001304430.1),点击“Fetch”直接下载至项目;
使用过滤器缩小范围(如限定物种“Homosapiens”或序列长度“>1000bp”)。
3.常见问题解决
格式不兼容:使用免费工具SeqManNGen转换文件格式,或通过“Edit”→“PasteSequence”手动粘贴原始序列;
大文件卡顿:在“Preferences”→“Memory”中增加缓存分配(建议≥4GB),并关闭实时预览功能。
二、DNASTAR 怎么加载序列
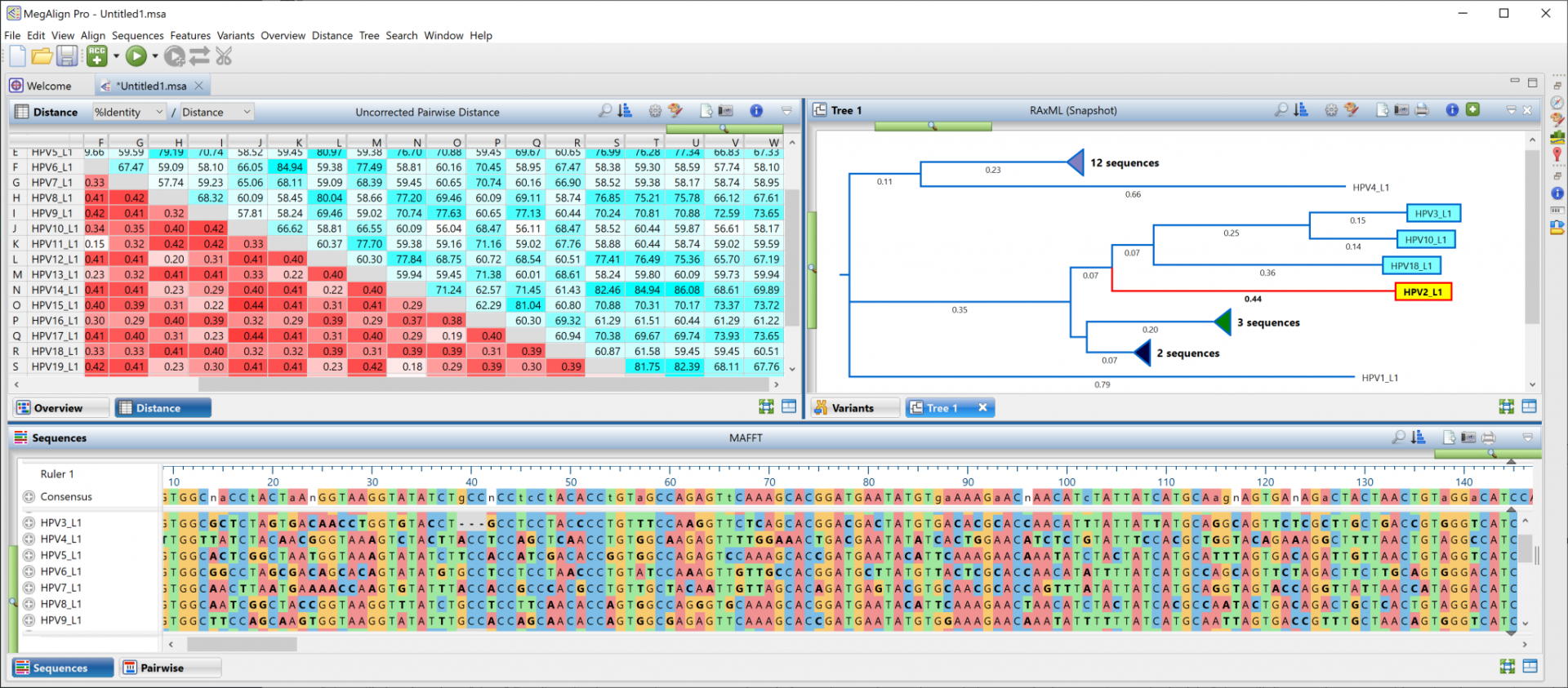
加载序列至分析模块是DNASTAR 进行比对、注释及可视化的关键步骤,需根据研究目标选择适配的工作流程。
1.单序列加载至编辑器
双击项目面板中的序列文件,自动在“SeqBuilder”或“GeneQuest”中打开;
使用快捷键“Ctrl+L”(Windows)或“Command+L”(Mac)快速定位特定碱基位置;
启用“互补链显示”(Show Reverse Complement)功能,对比双链结构。
2.多序列加载至比对工具
手动加载:
打开“Meg Align”模块,点击“Add Sequences”逐个选择文件;
调整序列顺序(拖拽文件名称)以优化比对结果;
批量加载:
创建“Sequence Group”文件夹,将同类序列(如同一基因家族)归组后整体拖入比对窗口;
使用“预设模板”(Template)保存常用参数(如ClustalW算法、空位罚分=10)。
3.动态数据加载技巧
实时链接外部数据:
在“Lasergene”套件中启用“Dynamic Link”,当原始文件(如Excel表格)更新时,DNASTAR 自动同步修改;
适用于长期跟踪实验数据(如突变株序列迭代);
云存储集成:
配置Dropbox或Google Drive同步路径,直接从云端加载共享序列;
设置权限控制(如仅允许团队成员编辑),保障数据安全性。
三、DNASTAR 多重序列比对自动化流程配置
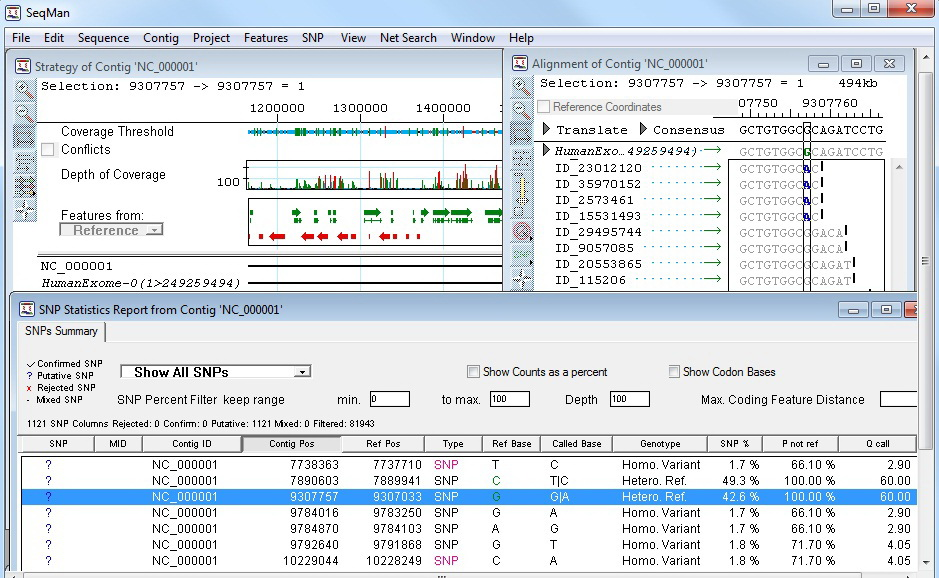
对于高频处理多序列比对的用户,通过自动化脚本和批处理功能可节省90%以上操作时间。
1.配置批处理任务
在“Tool Bench”模块中创建新工作流:
添加“Import Sequences”→“Alignwith Meg Align”→“Export Report”动作链;
为每个动作设置触发条件(如“仅当序列长度>500bp时执行比对”);
保存为“Batch_Alignment.dswf”模板,后续直接拖入文件即可自动运行。
2.脚本自定义进阶功能
使用内置Python脚本编辑器编写自动化逻辑:
应用场景:
高通量测序数据中批量剔除低置信度序列;
自动生成比对报告并发送至指定邮箱。
3.与第三方工具联动
通过“API接口”连接生物信息学平台(如Galaxy或Geneious):
将DNASTAR 比对结果导出为.nexus格式,供Phylogeny.fr构建进化树;
调用R语言脚本在DNASTAR 中直接绘制热图或PCA分析图。
DNASTAR 序列导入与加载的核心方法,并拓展至自动化比对的高阶应用场景。无论是基础序列编辑还是复杂多组学分析,合理利用这些功能可显著提升科研效率。建议结合DNASTAR 官方教程及社区脚本库,持续探索软件的深度潜力,以在分子生物学研究中保持技术领先优势。
